Laboratory Diagnosis of Antiphospholipid Syndrome
Article information
Trans Abstract
The diagnosis of antiphospholipid syndrome (APS) is essentially based on the detection of circulating antiphospholipid antibodies (aPL). Reading and interpretation of aPL profiles may be challenging and results of aPL should be interpreted in view of clinical context. The international diagnostic criteria of APS were developed and revised by international experts and societies, publishing international consensus guidelines on the recommended best practices for technical and performance requirements. Despite these attempts to produce consensus guidelines, some issues related to laboratory testing still remain unresolved. In this review, we want to give an overview of laboratory testing for APS with summary of recent international consensus guidelines for the detection of aPL. Also a brief review of prognostic significance of aPL profile and potential future diagnostic assay is presented.
Introduction
Antiphospholipid syndrome (APS) is a heterogeneous autoimmune disorder characterized by arterial or venous thromboembolic events and obstetric complications in association with the persistent laboratory evidence of antiphospholipid antibodies (aPL) [1-3]. As the clinical manifestations of APS lack specificity, the diagnosis of APS is essentially dependent on the detection of circulating aPL. These aPL are autoantibodies directed against a complex of phospholipids and phospholipid-binding proteins, not directly binding to phospholipids. aPL associated with APS include lupus anticoagulant (LA), anticardiolipin (aCL), and anti-β2 glycoprotein-I (aβ2GPI) IgG or IgM antibodies. LA is a subset of aPL that binds to phospholipid-associated proteins in coagulation complexes and disrupts phospholipiddependent coagulation tests [1-7]. aPL such as aCL and aβ2GPI antibodies are traditionally detected by ELISA, however newer automated platforms with various solid analytic and detection systems are now available in the diagnostic market.
The international classification criteria of APS were developed and revised by international experts and societies, publishing international consensus guidelines on the recommended best practices for technical and performance requirements [8-14]. To make a definitive diagnosis of APS, the presence of at least one clinical feature (vascular thrombosis or pregnancy morbidity) and one laboratory abnormality must be observed (Table 1). Persistent positivity of laboratory tests is important and the laboratory abnormality must be present on two or more occasions at least 12 weeks apart [9]. A remote test could avoid false positive results from the interfering events, but positive tests separated more than five years from a clinical manifestation also risk misclassification.
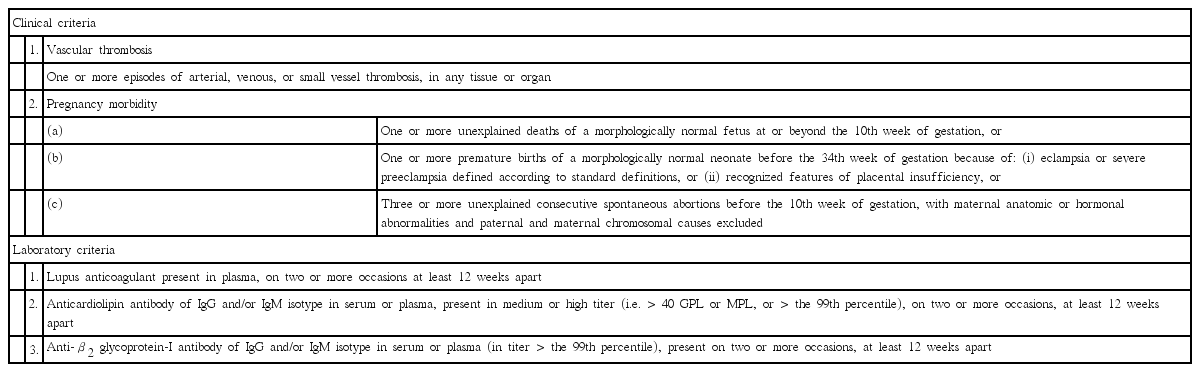
Summary of revised classification criteria for the antiphospholipid syndrome
Despite these attempts to produce consensus guidelines, some issues related to laboratory testing still remain unresolved. Poor assay reproducibility, variable sensitivity and specificity, and a lack of standardization and harmonization comprise methodological problems [6,7,15-17]. But progress has been made and reference calibrators for aβ2GPI immunoassays was newly developed [17]. In this review, we want to give an overview of laboratory testing for APS with summary of recent international consensus guidelines for the detection of aPL. Also a brief review of prognostic significance of aPL profile and potential future diagnostic assay is presented.
Lupus anticoagulant
LA is quite heterogeneous group of antibody and does not show uniform aPL activities, therefore laboratory detection of LA is limited to phospholipid-dependent clotting assays [1-3,6-8]. Consensus guidelines from different societies have been introduced and updated to guide the testing and interpretation process. In 2009, an update of the International Society on Thrombosis and Haemostasis (ISTH) guideline was published [10], and followed by a updated guideline from the British Committee for Standardization in Haematology (BCSH) in 2012 [11]. Recently, the Clinical Laboratory Standards Institute (CLSI) also published LA detection guideline in 2014 [14]. General agreements exist on sample preparation, use of dilute Russell’s viper venom time (dRVVT) in diagnostic repertoires, use of normalized ratios, phospholipid-dependent calculations, mixing test interpretation, and interpretative reporting [2,7,15,18,19]. Recommendations differ on reference interval cut-offs and all three cover testing of anticoagulated patients (Table 2).
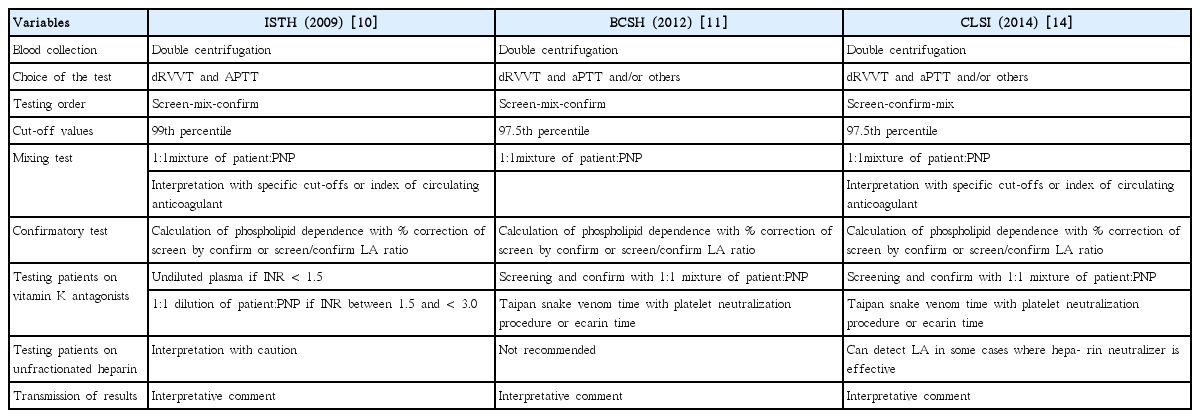
Summary of recent ISTH, BCSH and CLSI guidelines for laboratory detection of lupus anticoagulant (LA)
The current ISTH guideline stresses that LA testing should be limited to patients who have a significant probability of having APS, or who have unexplained prolonged activated partial thromboplastin time (aPTT) in the course of routine laboratory testing [10]. Double centrifugation to obtain platelet poor plasma is advocated by each guideline. There is evidence that no single assay system is sensitive enough to detect all LA and multiple assays are required to ensure weak LA detectable and to improve specificity, although one positive test is regarded as having a LA [9-11,14]. All guidelines agree on the use of dRVVT as the primary test for LA. The ISTH guideline recommends dRVVT for its specificity and aPTT with low phospholipid concentration because of its sensivity [10]. BCSH states that the second test can be an aPTT with proven LA sensitivity, a modified aPTT, or a dilute prothrombin time (PT) [11]. The CLSI guideline [14] recommends aPTT as the second test, but suggesting to employ additional tests. Recent studies using two different LA methods provided more objective LA reporting with additional diagnostic information [20,21].
Sensitivity and specificity of assays also depend on the cut-off values and can be improved by establishing local reference ranges. ISTH recommends the cut-off values above the 99th percentile of the distribution, whereas both BCSH and CLSI recommend 97.5th percentile of normally distributed data [9-11,14]. Mixing test is the second step in LA three-step procedure (screening, mixing, and confirmation) and improves the specificity. CLSI considers the mixing step as the last one and unnecessary in specific circumstances [14]. All guidelines agree that mixing tests should be performed with a 1:1 proportion of patient and pooled normal plasma (PNP). Test results are interpreted based on the cut-off value of clotting time or the calculated index of circulating anticoagulant (ICA). Confirmatory test must be performed by increasing the phospholipid concentration and in addition, BCSH suggest platelet neutralization procedure or LA insensitive reagent [10,11]. Phospholipid dependence is calculated as the percentage correction or LA ratio (screen/confirm) [15,18,19]. To improve the performance of LA assays, the conversion of clotting times for screen, mixing, and confirm tests to normalized ratio using the PNP value is advocated by ISTH and BCSH [1,2,10,11,15]. Integrated tests include screening and confirmation (low and high phospholipid concentration) in a single procedure on every patient irrespective of the abnormal screen test results.10,14,18,19 This approach is advantageous on the detection of weaker LA, but still raising concern on the performance of mixing tests [22,23].
All three guidelines concur that it is not ideal to test for LA while the patients are being treated with anticoagulant therapy [2,7,10,11,14,18,19]. ISTH recommends that LA test can be performed on undiluted plasma if the international normalized ratio (INR) is less than 1.5. Alternatively, if the INR is between 1.5 and <3.0, a 1:1 dilution of patient plasma and PNP can be considered [10]. BCSH and CLSI suggest LA test is possible on 1:1 mixtures with PNP for higher INR values [11,14]. ISTH and CLSI urge caution when interpreting LA results on patients receiving unfractionated heparin (UFH), whereas BCSH specifies that LA tests should not be performed, if the patient is receiving therapeutic doses of UFH, because it may cause erroneous results [11]. Only CLSI covers potential interferences by new anticoagulants, information on which is still awating [14,18,19].
LA results should always be considered in the context of a full laboratory aPL profile [10]. Three guidelines all state that quantitative results should be accompanied by an interpretative comment that indicating whether the findings are consistent with the presence or absence of LA [10,11,14,18,19]. Each supports the recommendation to retest at least 12 weeks apart for the evidence of antibody resistance.
Anticardiolipin and anti-β2 glycoprotein-I antibodies
In 2006, ISTH updated the clinical and laboratory criteria for APS [9]. According to the revised criteria, APS requires the presence of IgG and/or IgM aCL antibodies exceeding 40 IgG (GPL) or IgM (MPL) phospholipid units or 99th percentile, or IgG and/or IgM aβ2GPI antibodies at titers exceeding 99th percentile, on two or more occasions, at least 12 weeks apart. IgA isotype of aPL was not included in the current laboratory criteria. Both aCL and aβ2GPI ELISA should be performed following standardized procedures (Table 1). A new guidance from ISTH for the testing aPL with solid phase assays was recently introduced in 2014 [13]. To be useful in the standardization of the assays, it published ten recommendations covering from patient selection to results interpretation and reporting (Table 3). Various international workshops and the emergence of new platforms and technologies also have contributed to reduce laboratory variabilities [8,12,24-26].
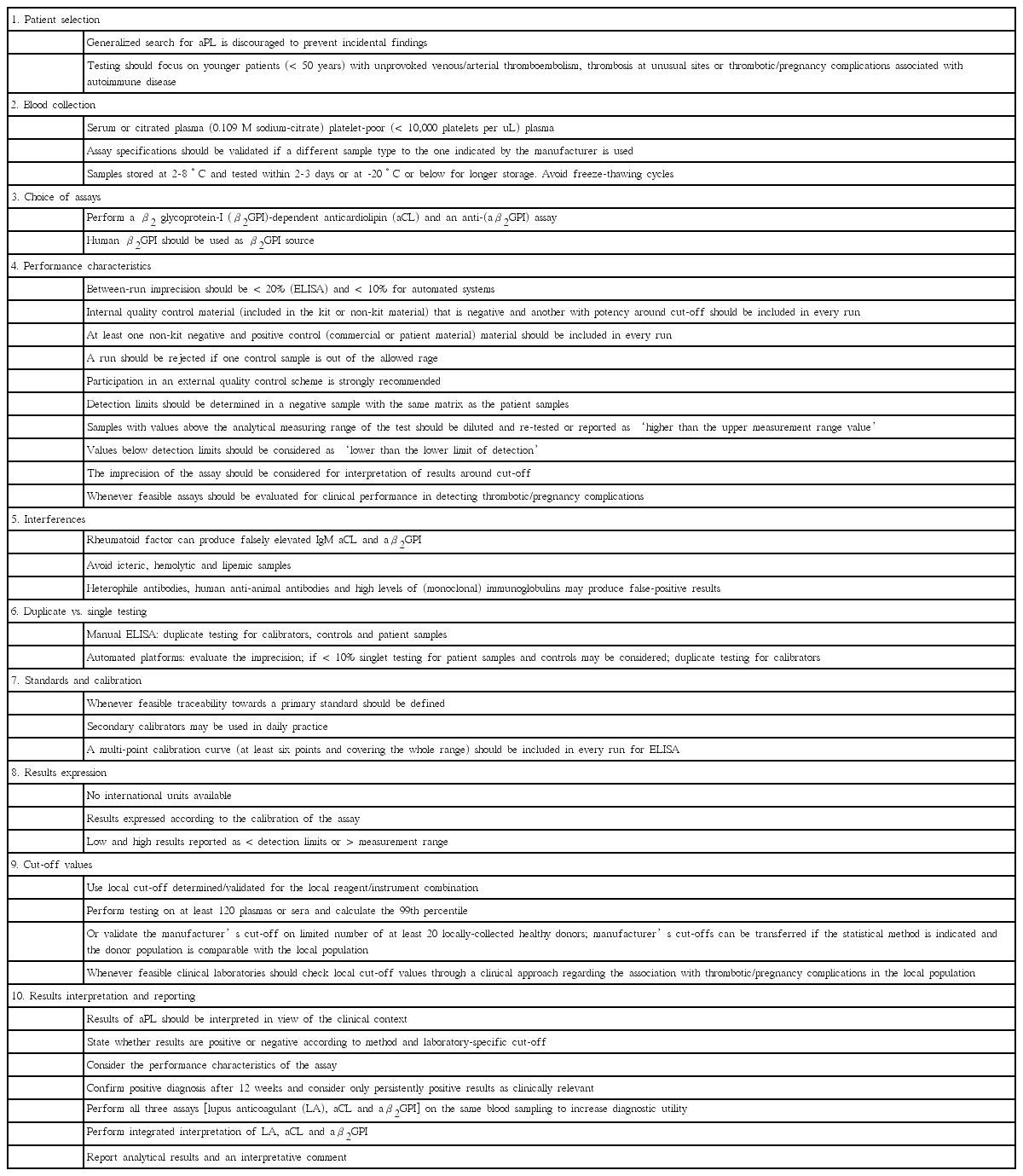
Testing for aPL by solid phase assays should also focus on patients who are likely to have APS [1-5,8,9,11-13,15]. Collection, storage and handling of samples are less critical compared to LA test. Serum or plasma can be used. The manufacturer should state the sample type recommended and plasma should be platelet poor by double centrifugation [12,13]. aCL antibody assays use the complex of cardiolipin plus β2GPI (human or bovine) as antigen. Because not all human aβ2GPI antibodies bind to β2GPI from other sources, whole molecule of β2GPI of human origin should be used [12,13]. As no gold standard exists, new assays should be validated before implementation. Linearity, precision, limit of detection, internal and external quality control, and clinical performance should be checked. The possible interfering factors such as cryoglobulins, rheumatoid factors, hemoglobin, bilirubin and triglycerides may cause bias on results [9,11-13]. To avoid errors, duplicate testing is recommended especially for manual ELISA tests, but technical progress with automated platforms may allow single testing for patient samples and controls. In contrast to ELISA, newer automated platforms does not require calibration curves for every run either, unless the reagent lot is changed [12,13].
The test signals are converted to antibody units derived from the calibration curve. Development of an international standard will facilitate the uniformity in reporting results of aCL and aβ2GPI antibodies [13,27]. Normal cut-off values should be determined in healthy subjects using the 99th percentile [9-13]. If this is not feasible, manufacturer’s cut-offs may be acceptable after adequate verification process [12,13]. aβ2GPI assay shows higher specificity than aCL for APS diagnosis and the use of 99th percentile cut-offs seem to be more sensitive than the >40GPL units [2,9,13,15]. Results of aCL and aβ2GPI antibody tests should be interpreted with LA results in view of clinical context. Each test result above cut-off should be regarded as positive and positive results need to be confirmed after 12 weeks. Inclusion of interpretative comment is strongly recommended [12,13].
Mostly aCL and aβ2GPI antibodies are measured by ELISA, although chemiluminescence (CLIA) and fluorescence enzyme immunoassays (FEIA) have recently been introduced to the market [16,17,27-33]. In most of the cases, fair to moderate agreement was found among different assays [16,17,29,31-33]. de Moerloose et al. [28] observed far higher agreement between three well-established commercial assays. However, on the contrary Gutensohn et al. [30] found moderate to poor accordance between five different assays in women with a history of miscarriage. The low agreement may be due to the selection of study population, low number of positive results, and different cut-off values. Different detection methods, antigen epitope variations, different assay designs, test interferences, and pre- or post-analytical issues all can be further reasons for the variability. Most samples showing imperfect agreement had values around the borderline or low-positive ranges. Utilizing an automated system can improve reproducibility and reduce interlaboratory variation [15-17,27-33]. Willis et al. [17] also analyzed that when the assays were calibrated using the recently developed reference IgG materials, the correlations for aβ2GPI antibodies were more improved.
Anti-β2 glycoprotein-I antibody against domain I
β2GPI is a 326 amino acid polypeptide synthesized by hepatocytes, endothelial cells, and trophoblasts. It contains five homologous domains of approximately 60 amino acids each, domain I at the N-terminus though to domain V at the C-terminus [1,2,24]. The β2GPI molecule circulates in plasma as a closed globular conformation and hides a cryptic epitope on domain I, thus preventing antibody binding. When β2GPI is immobilized on a negatively charged phospholipid surface via domain V, the configuration changes to an open form. This interaction induces exposure of the cryptic domain I to which aβ2GPI antibodies can then bind [1,2,8,24,34,35].
Recent studies demonstrated that aβ2GPI antibodies against domain I correlate well with thrombosis and obstetric complications compared to other domain-binding antibodies [1-3,5,8,13,15,24,26,34-37]. Because not all of the patients with aPL in their plasma suffer from thrombosis and/or pregnancy morbidity, it is suggested that there exist different populations of aPL. de Laat and de Groot [34] found that about half of patients with aβ2GPI antibodies showed reactivity toward domain I. Besides, the presence of anti-domain I antibodies showed better association with thrombosis at odds ratio (OR) of 18.9 and 95% confidence interval (6.8-53.2), compared with aβ2GPI antibodies toward other domains (OR 1.1, range 0.4-2.8). Through an international multicenter evaluation, they could confirm better correlation of aβ2GPI domain I antibodies with thrombosis and also found a better association with pregnancy morbidity (OR 2.4, range 1.4-4.3).
Mondejar et al. [36] and Pengo et al. [37] show that CLIA technology for aβ2GPI domain I antibody is more sensitive and reproducible than the existing ELISA assays. Antibodies that bind domain I of β2GPI appear to result in triple positivity (as defined by the presence of LA together with the same isotype of aCL and aβ2GPI antibodies), thus useful in identifying patients at high risk of developing thromboembolic events [34-37]. When we analyzed the prevalence of aβ2GPI domain I antibody in 178 samples that were referred for aPL test, 13 (7.3%) were positive for domain I, while 57 samples positive for one antibodor more test (Abstract for XXIXth International Symposium on Technical Innovations in Laboratory Hematology, Milano, Italy, 2016). Out of those 13 positive samples, 11 (84.6%) showed triple positivity. Overall, aβ2GPI domain I antibody test is very promising, however, further prospective multi-center studies are needed to help clarify the clinical utility of this new assay.
Triple anti-phospholipid antibody positivity
Current guideline advises investigators to classify APS patients into four different risk categories according to one or more positive aPL tests [9]. It is because certain issues of specificity and sensitivity of laboratory assays remain unresolved, while there are evidences that multiple aPL positivity is associated with increasing rate of thrombosis. Especially those with all three positive aPL tests (as triple positivity) are strongly associated with clinical events [2,3,5-7,11,12,24-26,38,39]. They showed increased risk of thrombosis and or pregnancy morbidity, with OR ranging from 5 to 33.
Several clinical studies indicate that triple positive APS patients are at high risk to develop recurrent thrombotic events [24,26]. Pengo et al. [38] also showed that 98% of initial triple positivity was confirmed after 12 weeks, while double or single positive cases were confirmed in 84% and 40%, respectively. Similarly during a two to four years of follow up for 136 initial positive samples, we could find that 100% of initial triple positivity confirmed aPL presence, whereas double or single positive cases confirmed positivity in 75.0% and 38.3%, respectively (unpublished data). Further study is required to determine whether aPL profile will affect APS classification and then influence clinical decision making.
Conclusion
APS requires the combination of at least one clinical and one laboratory criterion. Testing for aPL should be limited to patients who have a significant probability of having APS. Persistent positivity of laboratory tests is important. Solid phase aCL and aβ2GPI antibody tests should interpreted together with coagulation LA tests to assess the clinical significance. ELISA detection of aPL has been widely used domestically as well, although CLIA and FEIA methods have recently been introduced [31,33]. We also follow the revised international guidelines to monitor repeat positivities in Korea [40,41].
Several studies have shown that triple positivity correlates more strongly with both thrombosis and pregnancy morbidity than the presence of double or single positivity. IgG aβ2GPI domain I antibody was mainly present in triple positive patients and associated with thromboembolic events. However, we should be aware of interassay and interlaboratory variability and the performance characteristics of the assays.
Acknowledgements
This work was supported by the Dong-A University research fund.